Marco Galardini
26 April 2024
We are very happy to report that the first PhD thesis from the lab has been submitted this month!
With just one day to spare before the deadline (as it should be 😅), Dilfuza has submitted her
thesis to the ZIB office. Now we wait for the public defense in June.

In other news, last month Adam has officially left the lab to take an exciting new job as a postdoc
in the lab of Craig MacLean at the University of Oxford. Luckily Adam made a big push
before leaving and finished some large scale experiments thanks to his usual stamina, which will be
missed!

Congratulations to Dilfuza and Adam for these exciting news!
Marco Galardini
16 April 2024
As anticipated in the previous post,
we intended to run the Hannover half marathon, and indeed we did!
We all managed to get to the finish line, with a spatial mention to Hannes, who completed
the race in 01:59:52, with just 8 seconds to spare for the 2-hour psychological
barrier!

We didn’t quite manage to get a photo on the day of the race, but we are happy to report
that we collected 366 Euros through our DKFZ donation campaign. Thanks to all who donated!
Marco Galardini
25 March 2024
Our lab (Hannes, Judit and Marco) is running the Hannover half marathon on the 14th of April,
and we have decided that in order to boost our determination we needed support from colleagues, friends, and family.
And what better way to do it but to collect donations for a worthy cause? As researchers ourselves,
we have decided to support cancer research through the DKFZ (the German Cancer Research Center).
Cancer is the second cause of death in Germany, with an estimate ~270’000 deaths in 2019. Improved diagnostics and
therapeutics have reduced the death rates from cancer since the 90s, which clearly proves how a donation to cancer research has
the potential to affect the lives of many patients. So please help us get through the last two weeks of training and
through race day by supporting Cancer research! Follow the link below and scroll to the bottom of the page to find
the donation button. Thanks!
The Microbial Pangenomes Lab is running a Half Marathon for Cancer Research - DKFZ
P.s. if you are having trouble getting through the payment system do get in touch with Marco, and he can make
the donation on your behalf and collect the money afterwards through a bank transfer.
Marco Galardini
09 November 2023
Just a couple of days ago we have published a research article describing
panfeed, a software tool
to aid bacterial GWAS studies. This effort was
led by Hannes, with the help of Dilfuza
during peer review.
As everyone in the field of genomics has heard ad nauseam, we now have an abundance of
genome sequences available; when that is combined with phenotypic measurements the obvious
question is then “which gene is responsible for this phenotype?”. Statistical genetics (i.e.
genome-wide association studies, GWAS) would be one way to answer that question, or rather
the more correct one “which genetic variant is associated, and hopefully causal, for the
variation in phenotype, across this collection of genomes?”.
When asking this question in the context of bacterial genomes, the term “genetic variant”
can take a number of meanings, going from “classical” short variants such
as SNPs/InDels, to entire operons being transferred horizontally and even genes changing
their arrangement. One clever way to
encode all these variant types is to use a k-mer, which is a DNA “word” of length k
(typically 31).
While this solution allows one to collapse different genetic variant types into a unified
data structure, it generates two problems: one of ambiguity and one of interpretability.
These are the problems that we aim to ameliorate (hopefully solve for most people!) with panfeed.
Let’s go over both problems then.
Greatly simplyfing, one way to generate k-mers from a set of genomes is to take the input genomes
and put them into a blender that chops them into k-mers. The output can be then seen as a big haystack
from which we have to find the proverbial needle (the causal variants). The problem is that some genes
(which in bacterial genomes are pretty much the main unit of molecular function) will share some k-mers,
which will then implicate unrelated genes with the focal phenotype (as well as subtly modifying the presence/absence patterns
of those shared k-mers, affecting the association results themselves). That’s the ambiguity problem that needs to be solved;
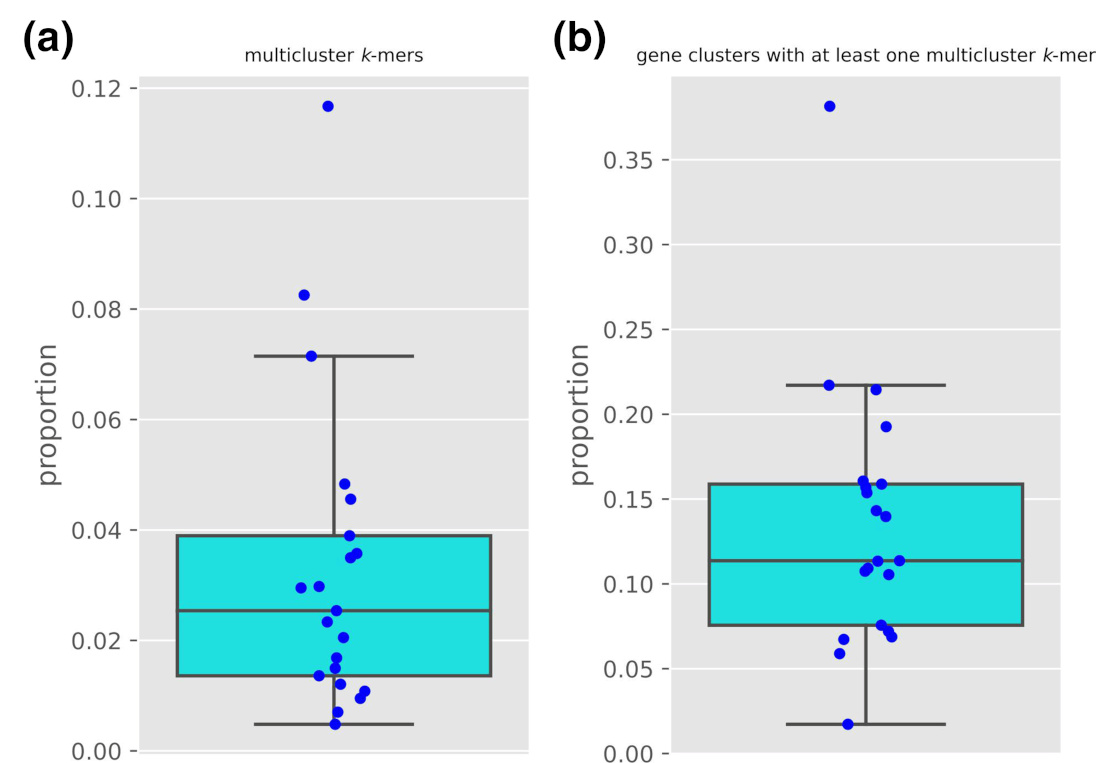
using data from 21 bacterial species’ genomes we indeed show how on average 3% of k-mers are duplicated across genes
and how 13% of genes have at least one k-mer shared with at least another gene.
The problem of interpretability also stems from all genetic variants being in a big haystack: once you have found your needle,
how do you figure out 1) which gene it might belong to, and 2) which variant type it encodes?
You can surely map the k-mers back to the input genomes, but you would lack the local context, meaning which other
genetic variants are present in that genomic location across all the genomes in the sample.
We address both problems by leveraging the assumption that genes are the unit of function in bacterial genomes and what
most people will use to interpret results. So basically instead of having a single haystack, we generate many (tens of thousands in fact!)
smaller ones and search for a needle in each of them. To be more specific, we use the gene cluster definitions given by tools
such as panaroo and ggCaller
and extract k-mers and their presence/absence patterns from each gene cluster separately.
This means that a k-mer that is duplicated across gene clusters is allowed to have a different presence/absence pattern for each gene,
thus reducing false gene leads.
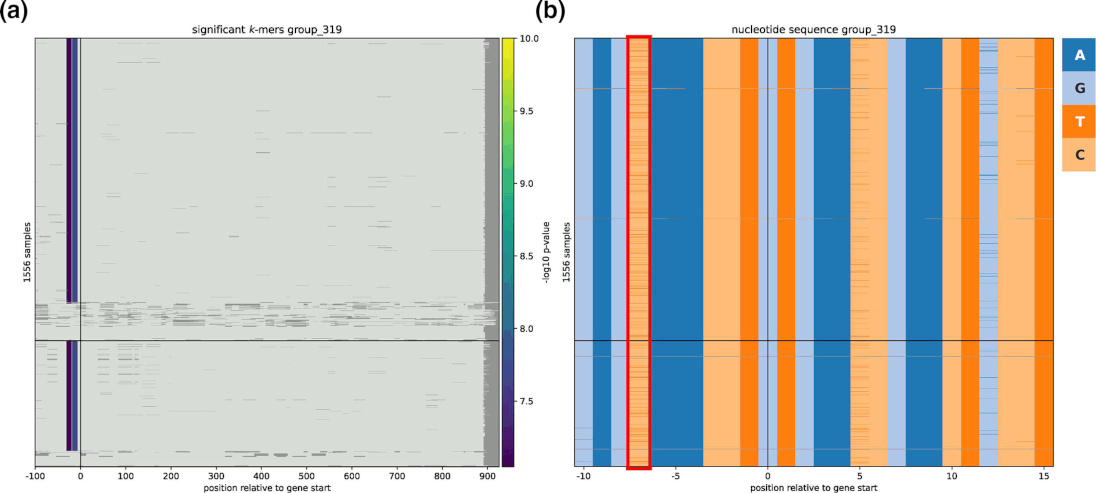
Interpretation is improved by linking k-mers and their source gene directly, as well as showing the local context of the associated variants,
which can optionally include promoter regions, as shown below.
panfeed is available as a conda package in bioconda (mamba create -c bioconda -n panfeed panfeed),
as a pypi package (python3 -m pip install panfeed),
and its source code and issue tracker can be accessed in our lab’s github. Happy needle search!
Marco Galardini
23 August 2023
Even though the lab is focused on bacterial pathogens, we (Marco and Gabriel)
took a look at the “2020 main character” pathogen, SARS-CoV-2, and just
put out a preprint on the topic of real-time identification of epistatic
interactions.
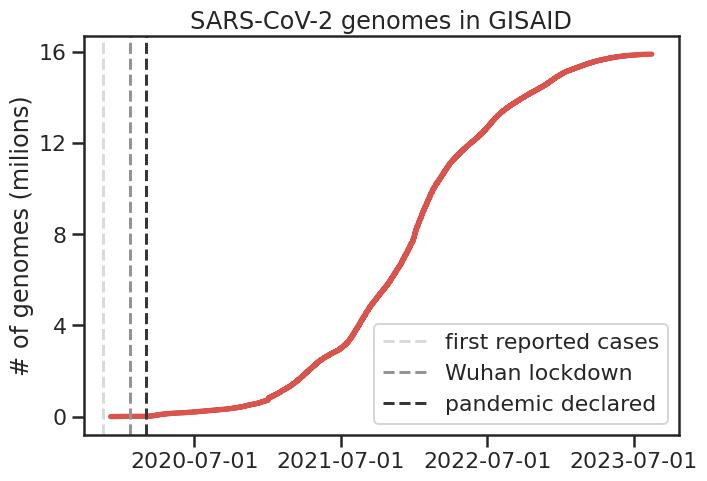
A particularly interesting development during the pandemic has been the
unprecedented amount of genome sequencing, and its use in very quickly
developing diagnostics (e.g. PCR primers) and treatments (i.e. sequence-based
vaccines and their updates).
Other applications closer to our field of study are the identification of
virus variants with a fitness advantage
and predicting the impact of genetic variants on genes and viral fitness.
We asked ourselves: can we find another use for this genomic data?
The application we were looking for would ideally be computed quickly, so that
it could be part of the public health decision process, and should leverage
the metadata associated with each sequence so that context would be taken into account.
We decided to look at a generally overlooked aspect in the evolution of this virus:
epistatic interactions. Generally speaking, two positions in the genome can be said to
interact epistatically if the impact of a double mutation deviates from the “sum” of the
effect of the single ones.
A particularly interesting example of macroscopic epistatic interaction can be found in the history
of scurvy.
We already know that these interactions are important for the evolution of
the Omicron variant (BA.1), has it has been elegantly showed through
experimental work.
These experimental studies are however relatively slow to perform and restricted to
certain regions of the genome (e.g. the Spike RBD). Could we come up with a
fast computational method that scales to millions of sequences?
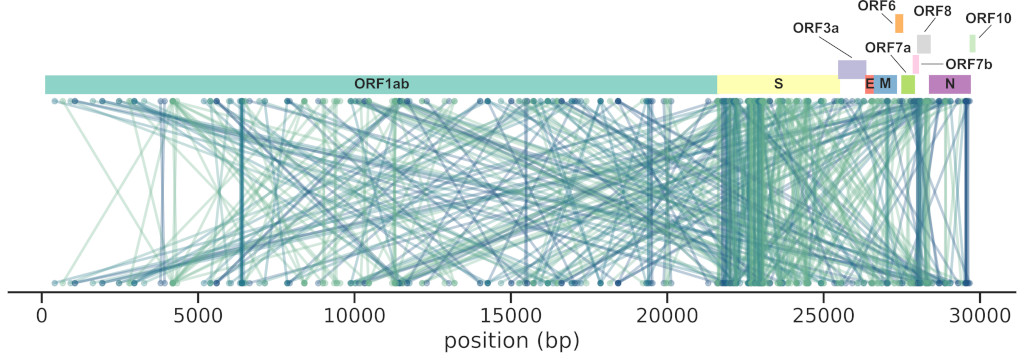
We reworked a method based on mutual information (MI) and applied to a large
set of SARS-CoV-2 sequences (~4M), finding 474 interactions across the genome,
the majority in the Spike gene (185).
Even though we made the method somewhat scalable, it took ~36 hours to complete
using several nodes on HZI’s cluster. But luckily we can obtain similar results
with as few as 10k sequences (a bit better if closer to 100k), which only takes
2 hours to complete.
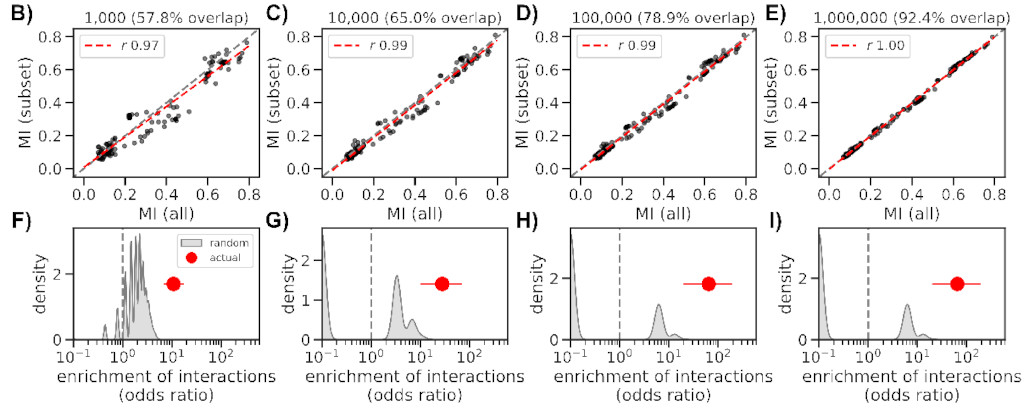
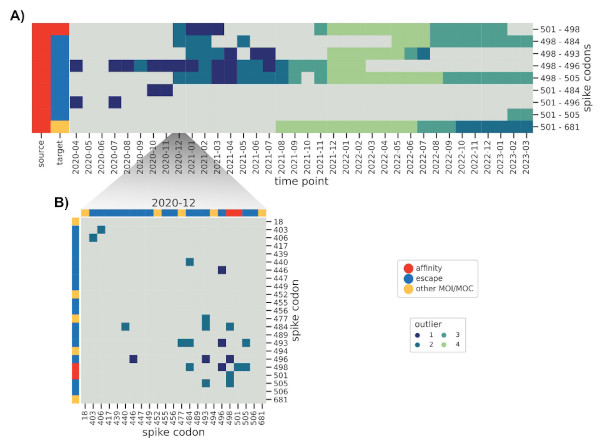
How do these interactions change over time, and how quickly can we spot known ones,
such as those found in Omicron BA.1? We further modified the method to reduce the influence
of older sequences and thus highlight emerging interactions.
And indeed we were able to identify a cornerstone epistatic interaction in the Spike protein
between residue 501 and 498 as early as 6 (!) Omicron sequences were present in the dataset,
which speaks to the sensitivity of the method.
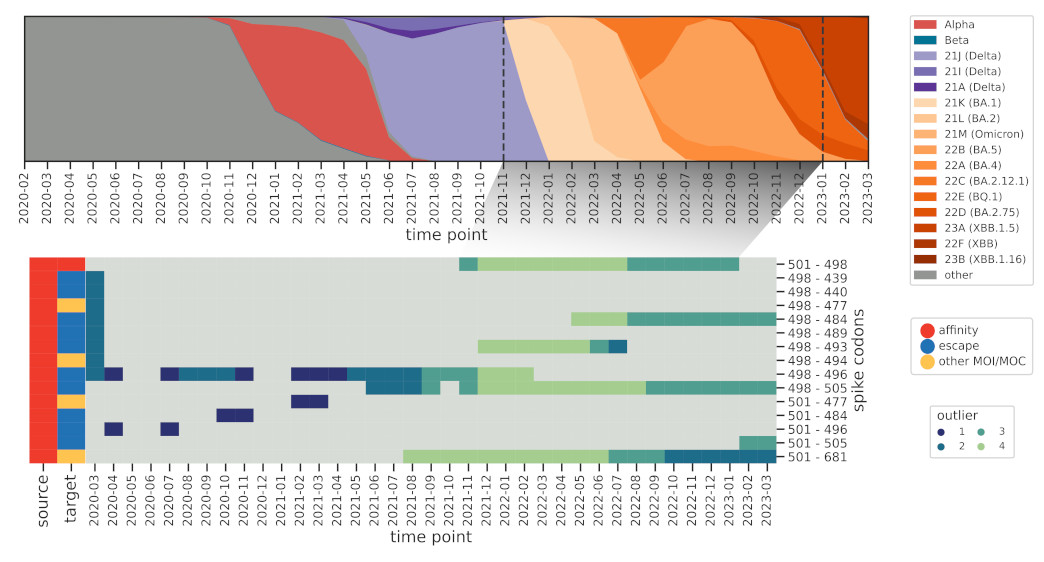
The flip side of this sensitivity is that genomes with incorrect metadata (i.e. date)
will make epistatic interactions appear at odd times, which was the case before we
used the excellent community resources put together by Emma Hodcroft.
We hope this work demonstrates that pathogen genomic sequencing is here to stay,
as the data can be used to generate many useful predictions that can in turn guide
public health decisions.
This work was started during the first lockdown, but it was significantly advanced by
Gabriel Innocenti, who was supported first by a FEMS research and training grant
and then by RESIST. A real pleasure to work with him!
P.S. as with our other papers, we have also shared the analysis code so that the study can be reproduced.